This HPLC evidence, corroborating the authentic structures, was further supplemented with experiments using dabsyl chloride derivatives of authentic preparations of beta-alethine and of authentic vitalethine, before and after heating.
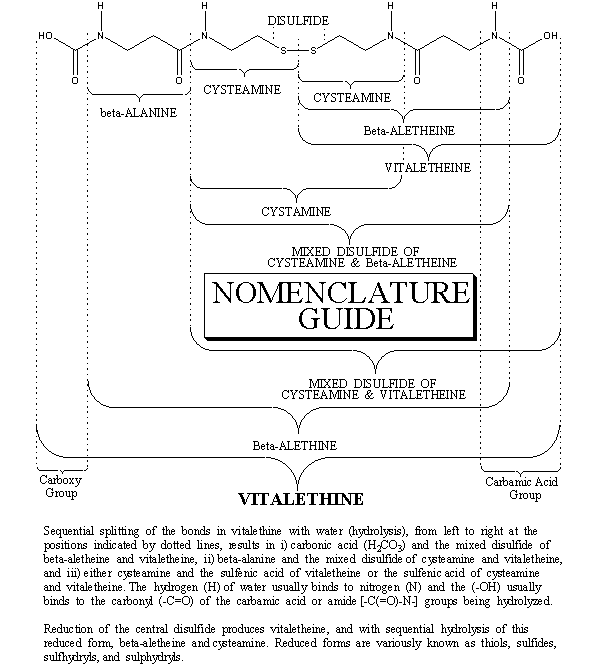
Heating beta-alethine is not likely to do much more than drive off the hydrogen chloride salts of the amines, thereby making the free amines more reactive with the dabsyl chloride reagent. When heated, an authentic preparation of beta-alethine produces a dabsyl amide derivative that binds fairly tightly to a reverse-phase HPLC column, eluting in a skewed peak late in the profile (37-43 minutes), long after the reagent peak elutes at about 24 minutes. The dabsyl amide derivative of cystamine elutes in the vicinity of the reagent peak. Note that there is virtually no evidence for the dabsyl amide of the amino acid, beta-alanine, in the heated authentic preparation of beta-alethine.
HEATED BETA-ALETHINE DABSYL AMIDE ELUTION PROFILE
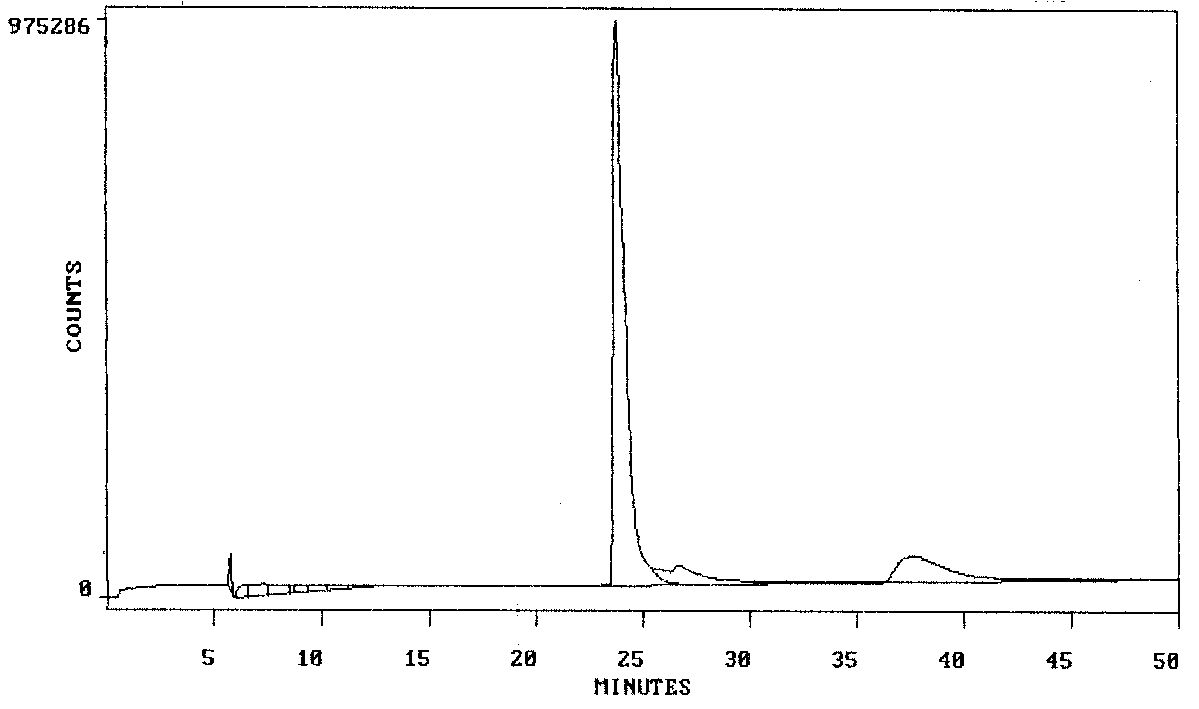
There also is virtually no evidence of carboxy-containing derivatives
eluting at the solvent transition (5 to 10 minutes) in the dabsyl amide
derivative of the authentic preparation of beta-alethine. This is as expected
for a relatively pure preparation of beta-alethine. The tiny peak eluting
at about 6 does raise some concern that the authentic preparation of beta-alethine
could contain traces of vitalethine or the amino acid, beta-alanine, the
carboxy group of either causing this compound or derivative to be much
more soluble in water, than the dabsylamide derivatives of the free amines
lacking carboxy groups. It is important to note that compounds containing
the hydrophilic carboxy groups, such as i) vitalethine, ii) a dabsyl amide
of the mixed disulfide of vitaletheine with beta-aletheine or cysteamine,
and iii) the dabsyl amide of the amino acid, beta-alanine, are NOT expected
to be retained much by the reverse-phase column, and should elute in just
a few minutes after injection onto the chromatographic column.
When authentic vitalethine is subjected to the same chromatographic treatment prior to heating, there is virtually no evidence for the presence of the dabsyl amides i) of beta-alethine (at 37-43 minutes), ii) of a mixed disulfide of beta-aletheine and cysteamine, or iii) of cystamine. Curiously, the reagent peak, 23 to 24 minutes, seems to be largely gone in this analysis, there being just three pronounced peaks, all in the 4-15 minute portion of the elution profile. Although not yet fully established by careful controls i) the relatively weak peak eluting at about 4 minutes could be underivatized vitalethine eluting in the solvent transition, ii) the sharp peak eluting at about 7 minutes could be traces of the dabsyl amide of beta-alanine caused by some decomposition of vitalethine under the conditions of the assay, and iii) the strong, tapering peak eluting at about 8 minutes might be unreacted dabsyl chloride reagent that was eluted from the column by the unreacted vitalethine. Alternatively, the reagent could have co-eluted with the dabsyl amide of a mixed disulfide of vitaletheine with beta-aletheine or with cysteamine, or a combination of the dabsyl amides of both mixed disulfides. A partial decomposition of vitalethine to dabsyl amides of beta-alanine and the mixed disulfide of vitaletheine with cysteamine under the conditions of this assay is the most likely explanation for the observed results.
ELUTION OF UNHEATED VITALETHINE + DABSYL CHLORIDE
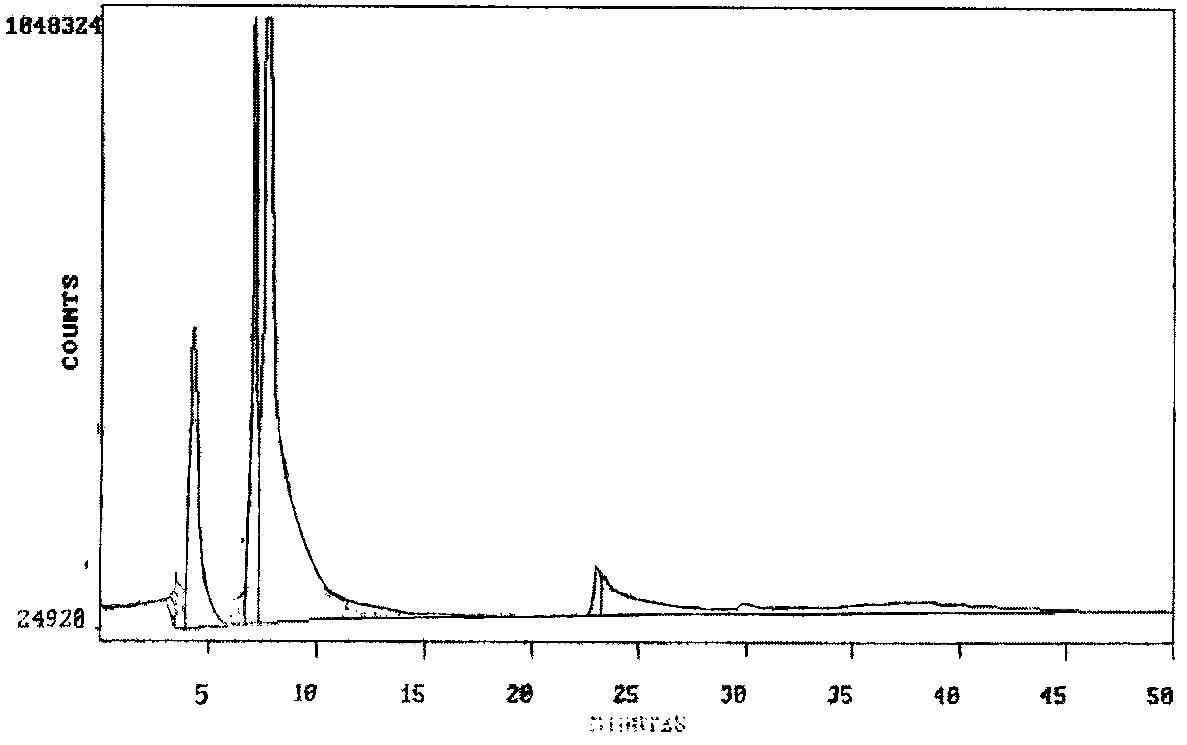
Early elution of much of the unreacted dabsyl chloride reagent from the reverse-phase HPLC column by vitalethine or with the dabsyl amide derivatives of mixed disulfides of vitaletheine with beta-aletheine or cysteamine, or both, is made feasible by the observation that many organic solvents seem to form inclusion complexes with members of the vitaletheine modulator family. In some cases, say with ethyl acetate, the tenacity of the solvent inclusions were graphically illustrated by an almost crystalline appearance which completely disappeared as the solvent was pulled off under slight vacuum and a steady stream of air.
Heating is an effective way of decomposing the carbamic/ carbonimidic acid moieties, whether they are neutralized to the carbamate/ carbonimidate salts with Ca(OH)2 or ZnO or not. Ammonium carbamate is much more stable in aqueous solution if neutralized with ZnO and recrystallized, presumably as the zinc carbamate, but this carbamate will still thermally decomposes before the water boils.
Heated vitalethine gave obviously different dabsyl amide derivatives compared to attempts to derivatize authentic vitalethine and the observed derivatives for heated preparations of authentic beta-alethine. As expected from the foregoing tentative assignments and discussion, the peak suspected of being unreacted vitalethine almost completely disappears upon heating, this peak shifting dramatically into a strong peak eluting at 7 minutes, again, suspected of being the dabsyl amide of the amino acid, beta-alanine. Consistent with this proposal is a dramatic increase in a new peak eluting at 32-35 minutes which is intermediate between the elution times expected for the dabsyl amides of cystamine and the dabsyl amides of beta-alethine. Thus, it appears that heating causes a disproportionate decomposition of vitalethine, the authentic compound decomposing to the dabsyl amides of a mixed disulfide of vitaletheine with cysteamine and of the amino acid, beta-alanine. These results are strikingly different from the elution profile obtained for the dabsyl amides of heated beta-alethine.
HEATED VITALETHINE DABSYL AMIDES/REAGENT
CONTROL
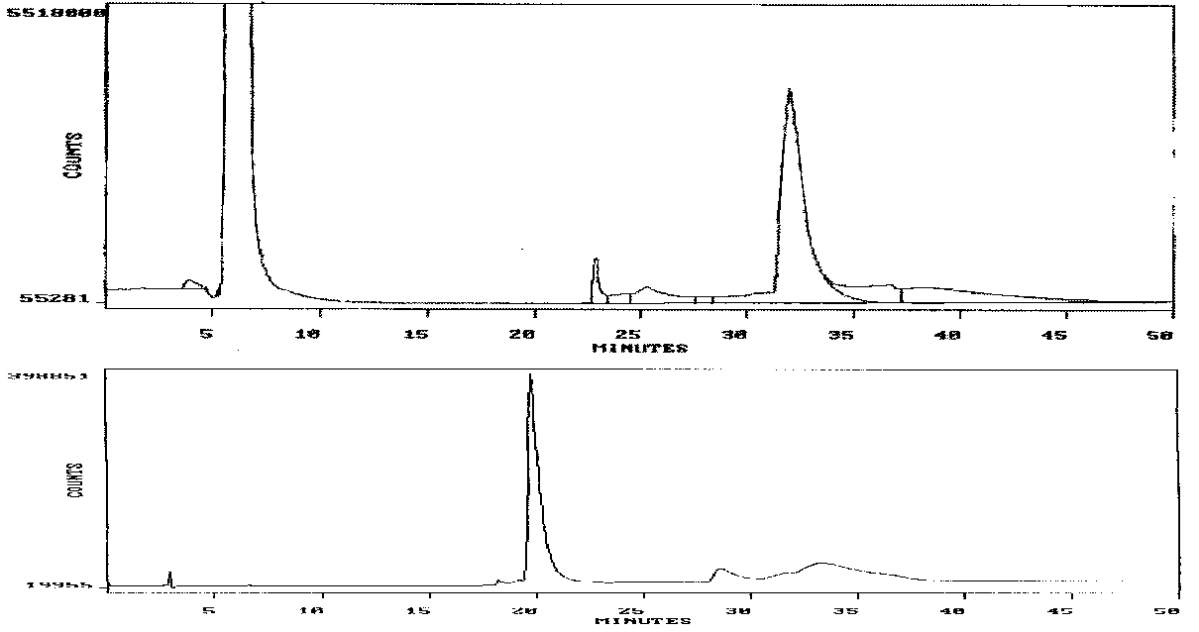
Note the slightly earlier elutions of the solvent transition (3-4 minutes) and of the reagent peak (about 23 minutes) in the control profile (bottom panel) relative to those in the chromatograph for the dabsyl amide of heated vitalethine (top panel), supra.
While others might argue that the vitalethine tested was not pure and that it was largely contaminated by a mixed disulfide of vitaletheine with beta-aletheine or with the amino acid, beta-alanine, such allegations would be inconsistent i) with the relative absence of these accompanying decomposition peaks until after heating and ii) with the beautifully clean 13-C and 1-H NMR spectra obtained for unheated, authentic vitalethine. An unpure preparation of vitalethine, contaminated with these decomposition fragments, would have exhibited many more methylene peaks in the carbon and proton NMR spectra, instead of just the four methylene peaks observed in each of the NMR analyses.
The only conclusion that can be drawn from these definitive experiments is that vitalethine cannot be beta-alethine. Vitalethine even thermally decomposes in a different way than authentic preparations of beta-alethine. Under less complicated conditions with regard to the chemical environment, the IR spectra of authentic vitalethine and of authentic preparations of beta-alethine before and after heating indicate that vitalethine can thermally break down into a product that closely resembles the thermal decomposition product of authentic preparations of beta-alethine. Thus, though related, the distinct chemical and biological differences under ambient physiological conditions between the authentic preparations of beta-alethine and the authentic vitalethine are undeniable at this time.
GO TO:
| Home | Overview | People | Journal | Nutrition |
| Environment |
|
WWW Links | Outline | e-mail us |