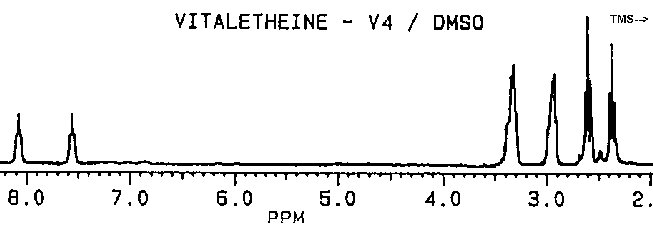
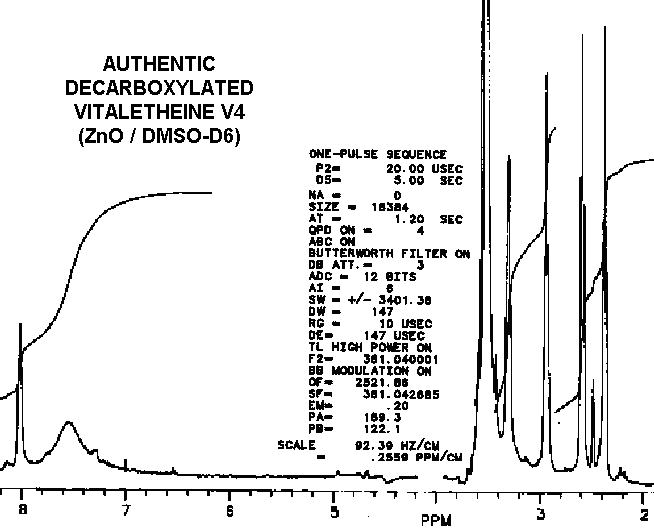
The coupled proton NMR spectra of vitaletheine V4 in DMSO is particularly interesting since the coupling and amplitude of the N-H and adjacent methylenes appear to be nearly identical with the undeblocked compounds. This would not have been the case if decarboxylation to the free amine had occurred, since removing the carbon dioxide from the carbamate moiety should approximately triple the number of observed protons, as the free amine (protonated at physiological pH) is formed from the carbamate. Concomitant with a tripling of protons on one of the amines upon decarboxylation, the methylene quartet at about 3 ppms should change as the number of readily exchangeable protons on the adjacent amine increases. Both, an increase in amino protons and a transition from a quartet to a broad triplet, have actually been observed as authentic vitaletheine V4 is decarboxylated in DMSO. Since DMSO theoretically can draw Zn ions out of the vitaletheine V4 complex, the stability of vitaletheine V4 in DMSO that has not been saturated with zinc or other stabilizing cations is somewhat questionable at this time. Although it is clear that these spectra provide irrefutable evidence for the carbamate structure in vitaletheine V4-like compounds, how extensively this particular preparation was exposed to anhydrous and hypobaric conditions is not known. These conditions theoretically can remove ZnO from the complex, resulting in an imidate anhydride with the carbonimidate moieties still intact.
With ample evidence for a carbamate in its proton spectra, the lack of an obvious carbonyl carbon in other's carbon spectra of vitaletheine V4 is troublesome. Possible explanations for this include i) dispersion of the signal for the carbon of the carbamate moiety over two possible ionization states and two possible resonances, including the downfield carbonimidate tautomer, ii) dispersion of the signal for carbons in an asymmetric polymethylenoxy ring over shifts for a variety of possible steric constraints in the spectra, and iii) an exact or nearly exact alignment of one or more of these carbon peaks with other peaks, such as the amide, for which there is some slight evidence. Differences in the relative intensities and positions of peaks are noted between the spectra of authentic vitaletheine V4 preparations and of those decarboxylated derivatives produced by others.
Even slight modifications to the published and exemplary procedures can produce compounds with NMR very similar to the vitaletheine modulators, but oxidation states higher than sulfenates (i.e., sulfinates and sulfonates) differ chemically from authentic samples made with the published procedures. These differences are made obvious when authentic benzyl derivative (zinc salt) is passed through a thiol exchange column in the presence of saturating amounts of zinc ions to reduce the sulfenate linkages. An underlying proton NMR spectra, nearly identical with authentic vitaletheine V4, is obtained in the partially-reduced effluent. Since these "reducing conditions" would not be expected to oxidize the sulfur to higher oxidation states, the most likely explanation for these results is that, upon reduction, the benzyl derivative polymerizes to vitaletheine V4 or a very similar derivative. Further reduction to the free thiols (zinc sulfides?) probably would be necessary for elution of the compound from the thiol and disulfide exchange column. Since there also seems to be some slight bifurcation in the proton NMR peaks representing the benzyl ring, reduction of the benzyl derivative on this column may also remove the benzyl group as the tetramer is formed.
Similar reductions of the bis-CBZ-ß-alethine starting material would normally have been performed to ensure that the observed spectrum of the reduced benzyl derivative is not that of CBZ-ß-aletheine. However, this starting material is insoluble in water, making such a comparison under identical conditions difficult. Logically, any CBZ-ß-aletheine in the effluent, even its zinc sulfide, should eventually autoxidize to the disulfide starting material and precipitate from the effluent (assuming, of course, that the intermediary sulfenates do not simply reform the sulfenate-linked dimer). When coupled with the observation that a published proton spectrum of CBZ-ß-aletheine bears little resemblance to this column effluent it seems unlikely that the material produced by this reductive process is CBZ-ß-aletheine. At the same time it is important to realize that the effluent could just as easily be the zinc sulfide instead of the free thiol and that the spectra of zinc sulfides often mimic the proton spectra of their corresponding disulfides. This may explain, in part, why the proton spectra of ß-aletheine is so very similar to the original disulfide, ß-alethine. As expected from this rationale, the proton NMR spectra of ß-aletheine (presumably the zinc sulfide) also lacks most of the fine spectral splitting attributed to hydrogen bonding in authentic ß-alethine.
These observations indicate that the benzyl derivative must be handled very carefully with regards to reductive conditions and absorptive materials such as sterilizing filters and column matrices. It is also apparent from these results that the process of making authentic vitaletheine V4 from their common precursor with UV irradiation is probably photoreduction and not the oxidation proposed by others. These observations, therefore, tend to support the proposed synthetic mechanisms and structures for authentic vitaletheine V4 and the sulfenate-linked benzyl derivative.
These considerations also bring up other interesting points. Nucleophiles are spaced in this series of compounds to facilitate five- and six-membered ring closures. Sulfur is an excellent nucleophile and such facile intramolecular cyclization through the carbonyl carbons of amides, imidates, carbamates, and carbonimidates theoretically should make thiolates and thiols of these compounds less reactive to alkylating and other derivatizing agents. These considerations help to explain why thiol derivatizing agents, such as the bromobimanes, are far less useful for characterizing thiols in this family of compounds, and why these authentic vitaletheine modulators have been mistaken as sulfonates by others.
Even amino derivatives of vitalethine are not obtained until the modulator is heated to thermally decompose the carbamate. Note that this exposure of the free amine to reagent only upon heating provides further support for the assigned carbamate/carbonimidate structures for vitalethine and vitaletheine V4. Thermal release of a gas that can be trapped as a barium salt (presumably the carbonate), and at temperatures far lower than those required to melt the benzyl derivative's calcium salt, also tend to support the carbamate/carbonimidate structural assignments for vitalethine and vitaletheine V4.
Additional analyses using proton and carbon NMR, including two-dimensional NMR, have helped to establish the carbon and proton assignments for vitalethine and beta-alethine. At the same time, this work helps to confirm the chemical differences between these two preparations, while, unfortunately, also illustrating just how important it is to do very careful side-by-side analyses of these compounds in any analytical approach.
GO TO:
| Home | Overview | People | Journal | Nutrition |
| Environment |
|
WWW Links | Outline | e-mail us |
{kind=link}
{kind=link}